5. Creating / modifying ligand molecules¶
5.1. Introduction¶
As mentioned in the introduction to Force Fields, we may be lucky and be able to download the structure of the ligand that we want to dock to a given protein target but in reality our research projects are likely to involve new molecules (or at least modified analogues of existing ones). In order to perform calculations with these we will need to do some building and geometry optimisation on our molecule.
There are various software programs in existence that will allow you to do this but in this practical we will make use of the free software Avogadro.
Most importantly for our purposes Avogadro is a powerful molecule editor/builder, however it is capable of much more. Detailed information on the software, its features and how they are used can be found in the Avogadro manual. Here we will focus on some of the most useful features within the context of preparing ligand molecules for docking calculations.
5.2. Getting started¶
Avogadro can be started from the command line in Linux by simply using the program name to start an empty Avogadro session:
avogadro
The Avogadro window should look like this:
Note
avogadro may look slightly different depending on exactly what operating system you are using e.g. in the most recent versions of Ubuntu Linux the default is a newer version with a slightly different interface layout: avogadro2. The previous version shown here is not installable as it requires the QT4 libraries and current versions of Ubuntu use QT5 which is not compatible. However, the main difference is that some functions like energy minimisation are accessed through the drop-down menues instead of the buttons shown here.
If you wish to open it with an existing molecule file, e.g. ligand.mol2, the filename follows the program name:
avogadro ligand.mol2
Here we have the Avogadro window with a paracetamol molecule loaded:
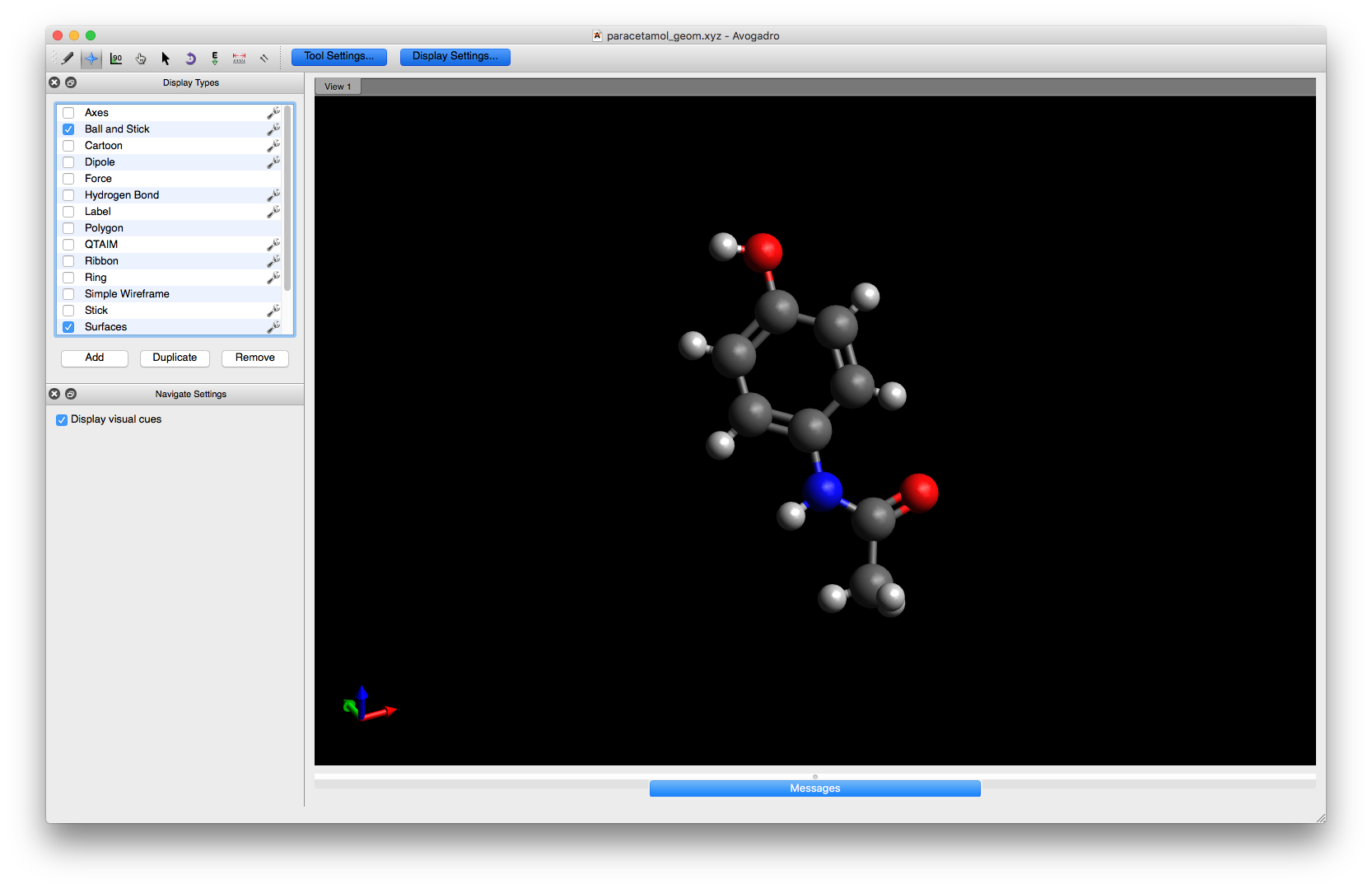
The coordinates for this paracetamol model (in XYZ format) are:
20
Paracetamol
O -0.22878 2.41641 3.08195
H -1.06168 2.75162 3.29313
C -0.29831 1.56695 2.00247
C -1.39580 1.55120 1.13925
H -2.14690 2.13474 1.30145
C -1.40503 0.69720 0.04926
H -2.17820 0.68794 -0.55158
C -0.32590 -0.15216 -0.20466
C 0.75629 -0.15309 0.67726
H 1.47460 -0.72922 0.53205
C 0.76447 0.70368 1.76980
H 1.51597 0.69905 2.37354
N -0.42796 -0.99412 -1.34282
H -1.27346 -1.08119 -1.70685
C 0.56135 -1.59341 -2.03055
O 1.75209 -1.51792 -1.70696
C 0.11451 -2.38906 -3.23180
H -0.75994 -2.13990 -3.46516
H 0.13418 -3.30512 -3.02501
H 0.69187 -2.21863 -3.95268
Copy these coordinates and save in a file called paracetamol.xyz and we will use them as a base to make some modifications.
With a molecule loaded you can manipulate and modify it using the tools accessible through the menus or the tool icons. For pop-up summaries of what the mouse buttons do in each tool mode over the mouse pointer over the menu buttons
5.3. Modifying paracetamol¶
Open your paracetamol.xyz file with Avogadro:
avogadro paracetamol.xyz
To add or change an atom go to the menu buttons at the top of the window and click on the pencil icon to open the drawing settings
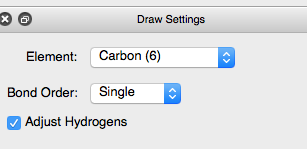
Select the element Carbon (6) and ensure that the Adjust Hydrogens box is checked. Then, click on the hydroxyl hydrogen with the left mouse button to change it to carbon. Hydrogens will automatically be added to satisfy the valency on the carbon atom and your molecule will now contain a methoxy group on the phenyl ring.
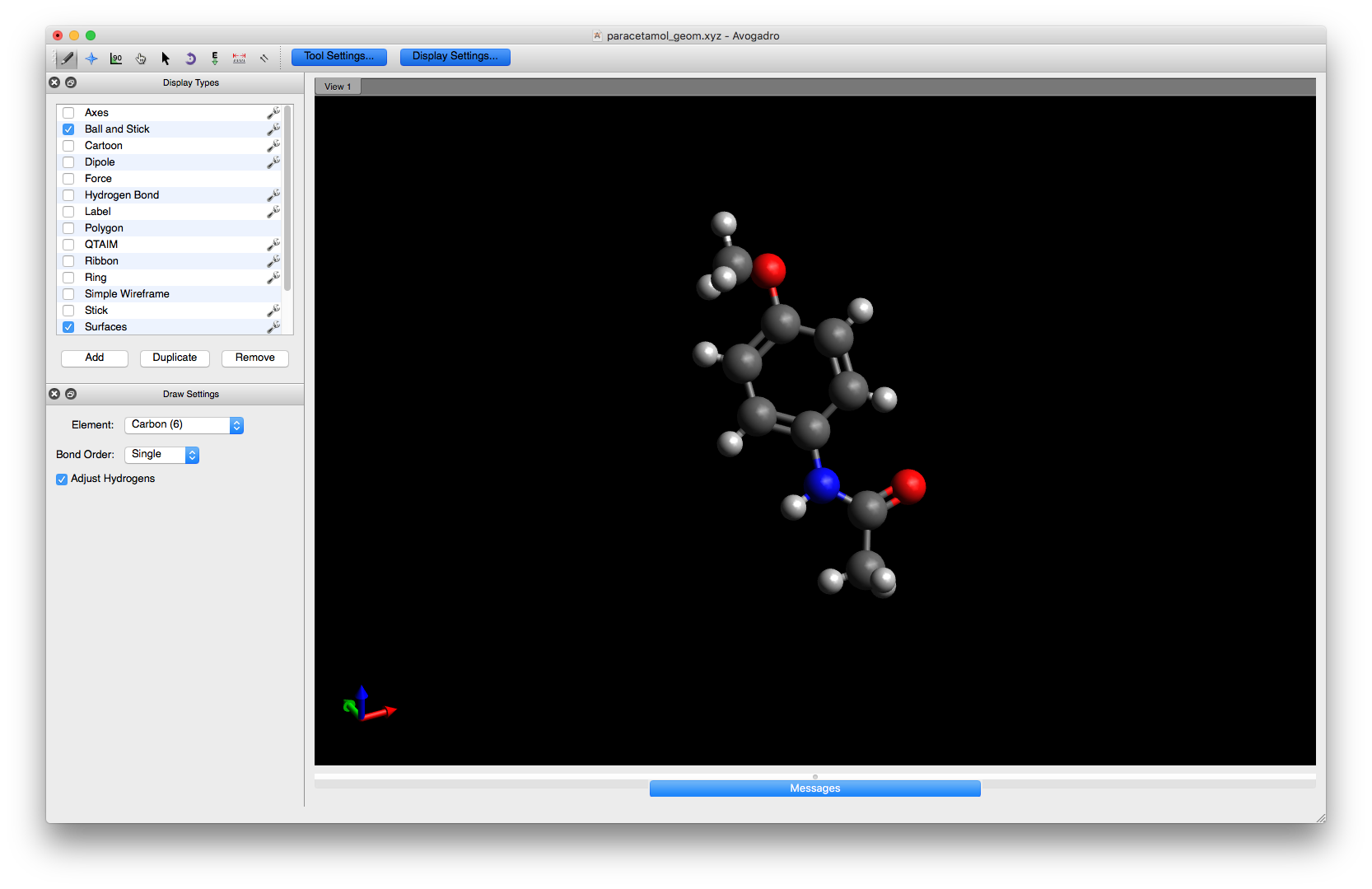
This molecule now contains a chemical modification but the geometry is not correct (Avogadro does not change the bond length, so the O - C distance is the same as the original O - H one which is clearly wrong). To fix this we must do an energy minimisation to obtain a more realistic geometry.
Click on the tool button with an E and a small arrow to obtain the optimisation settings dialogue and select the MMFF94s force field which gives good results for organic molecules. Press Start and you will see the geometry in the window changing.
As the geometry is optimised you will see the energy and gradient of the energy changing at the top of the screen. Once the gradient reached zero, the optimisation will be terminated. At this point it is a good idea to save the structure to a new file (File > Save as) such as methoxy_paracetamol.xyz.
Note
You will see that there are options in the Save dialogue to save in different file formats. This is possible because Avogadro is built around an older version of the command line software OpenBabel. In fact, the molecular mechanics energy minimisation routines are also provided by OpenBabel and could therefore be done on the command line without using the graphical user interface of Avogadro but we will not go into that here.
Whilst in the energy minimisation mode you can move the molecule around with the mouse:
Left button: rotate the molecule
Middle button: zoom in or out (you can do this with the wheel as well)
Right button: translate the molecule
Double click: reset the view
Warning
Be careful not to ‘grab’ an atom with the left mouse button as it then becomes captured and you will be able to drag it around during the optimisation.
Actually, this can be a useful tool if you want to change the geometry by hand but be careful as it is easy to introduce errors e.g. invert the stereochemistry at a chiral carbon.
5.4. Adding hydrogens¶
Another common task in ligand preparation is the addition of hydrogen atoms. There can be several reason for this:
If your ligand comes from a protein crystal structure it is unlikely to contain hydrogens as these structures usually do not have sufficiently good resolution to establish the hydrogen positions.
Similarly, if you download your ligand structure from an online database it is possible that it may not contain hydrogens (although to be fair, this is a less common occurrence then it used to be).
You may want to simulate the effects of pH on your ligand if it has titratable groups (for instance, if the protein target is located in the upper gastrointestinal tract the pH will be different to that found in e.g. lung tissue).
To do this you can use the Build menu and select either Add Hydrogens to add all possible hydrogens or Add Hydrogens for pH... and then select a pH for the protonation algorithm (usually you will use the default which is pH = 7.4 to ensure that your molecule is correctly protonated for physiological - i.e. intracellular - pH). Both of these functions are provided by the OpenBabel software built into Avogadro.
Note
If your molecule already contains hydrogens, when you select one of these options the existing hydrogens will be removed and replaced with the Avogadro/OpenBabel ones.
5.5. Building molecules from scratch¶
Sometimes you will want to generate a completely new structure and this can be done by using several of Avogadro’s build functions. We will build the paracetamol molecule from scratch as an exercise.
Start a new Avogadro session. We could start by drawing the phenyl ring by hand but instead let’s make use of the fragment database in the Build menu:
Build -> Insert -> Fragment...
This will bring up the Fragment selection tool
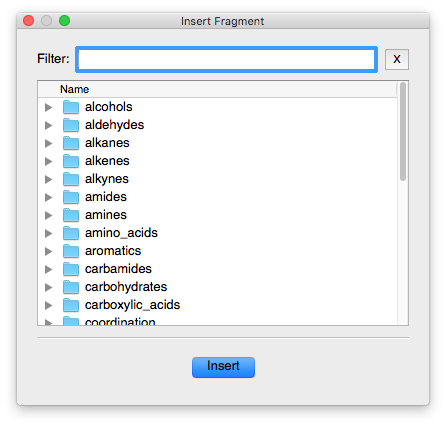
In the search/Filter box, type benzene, click on benzene.cml and press Insert. You will now have a benzene molecule in the main Avogadro window. Note, however, that all the atoms are selected at this point (they will be surrounded by transparent blue spheres)
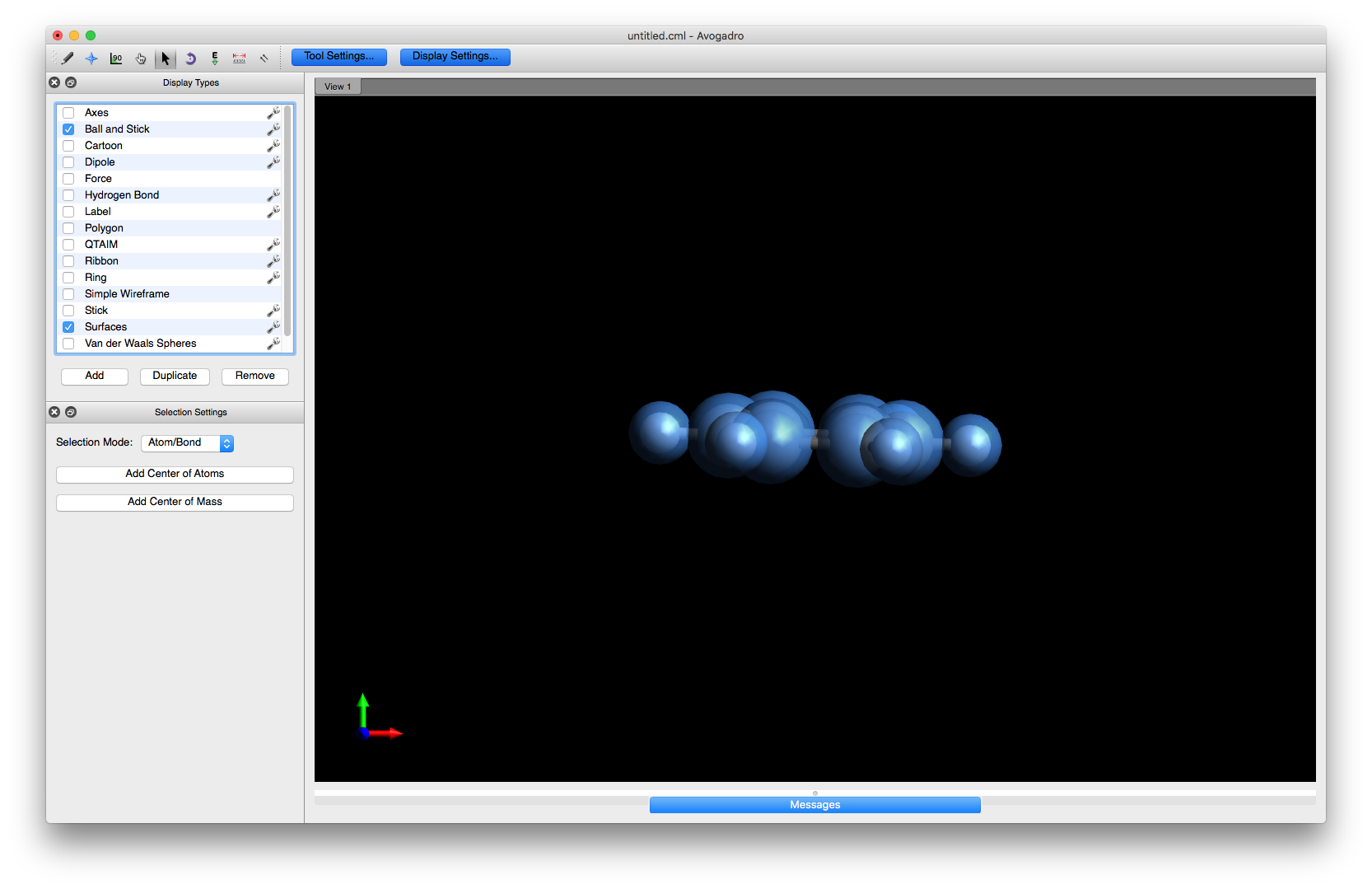
and you need to deselect them by choosing the Select tool button (which looks like the mouse pointer) and using the left mouse button and dragging somewhere in the main window at which point the blue spheres will vanish.
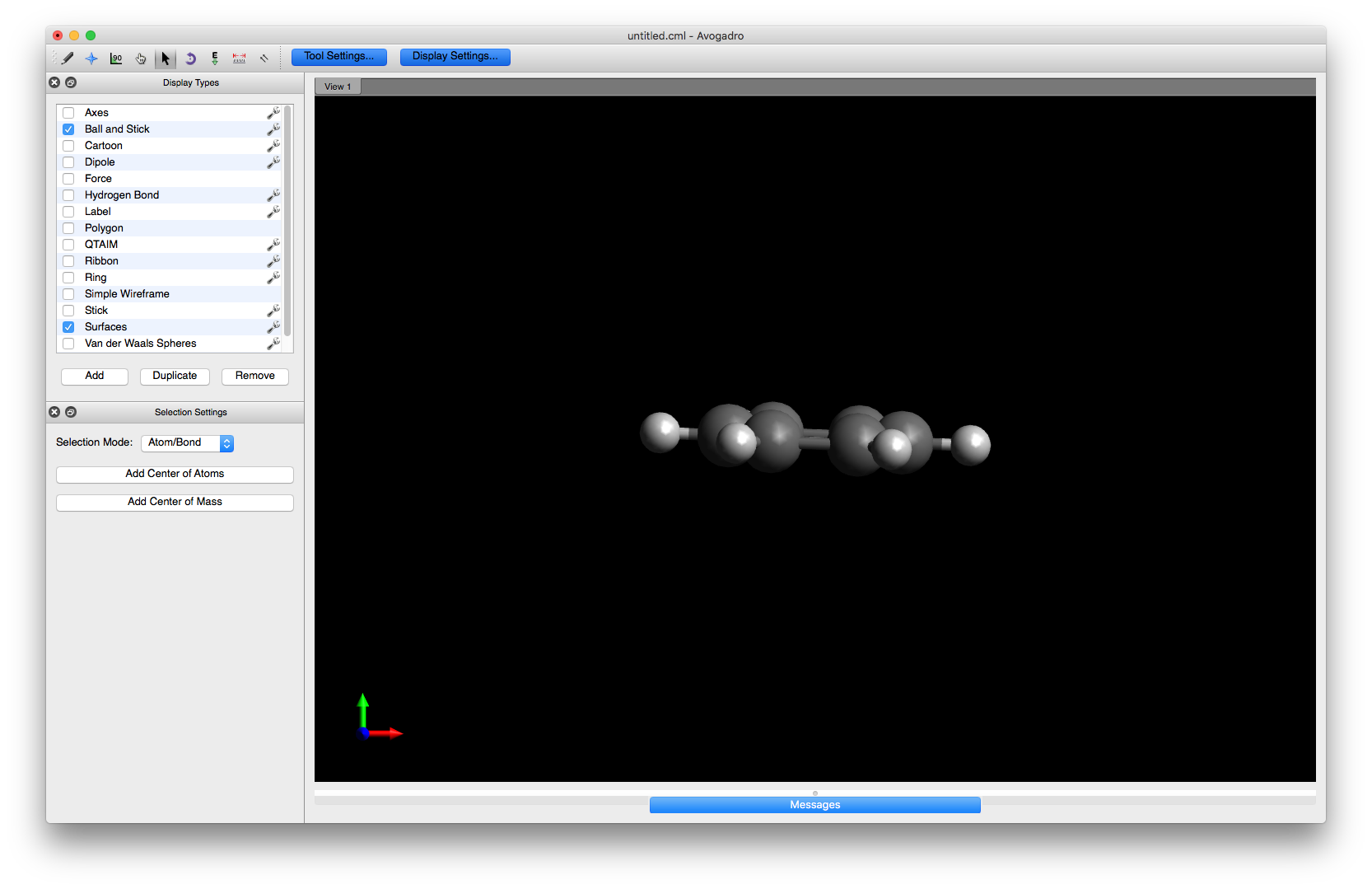
You are now ready to begin altering the structure. First, rotate the benzene molecule into an orientation that is easier to work with. You can either select e.g. the Manipulate tool and use the mouse or click in the main window and then use the keyboard arrow keys to rotate the molecule
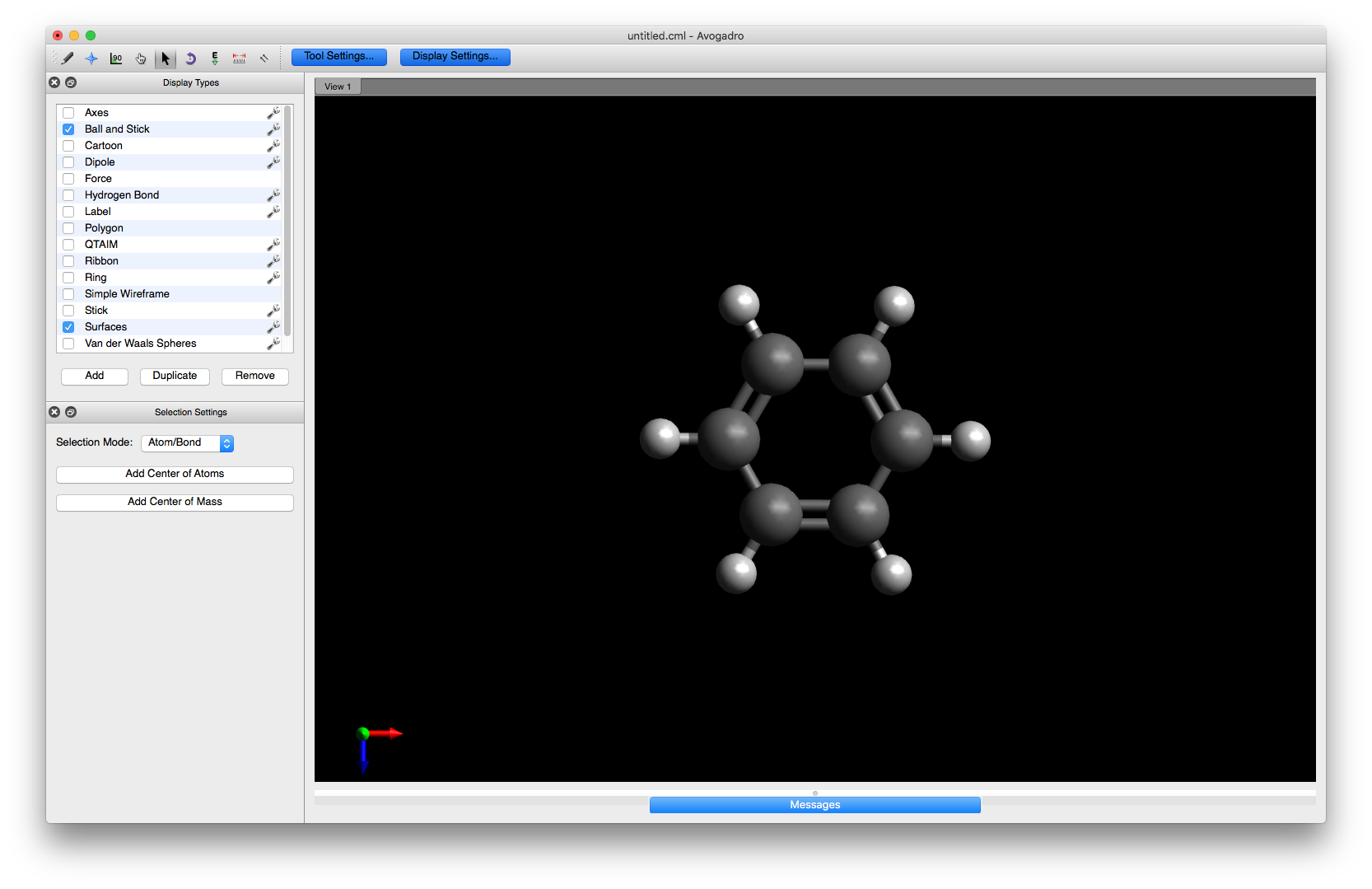
To begin editing the structure, choose the Draw tool and select Oxygen (8) from the drop down menu. Click on one of the hydrogens to change it to a hydroxyl group or alternatively, click on one of the carbons and drag the mouse a small distance away
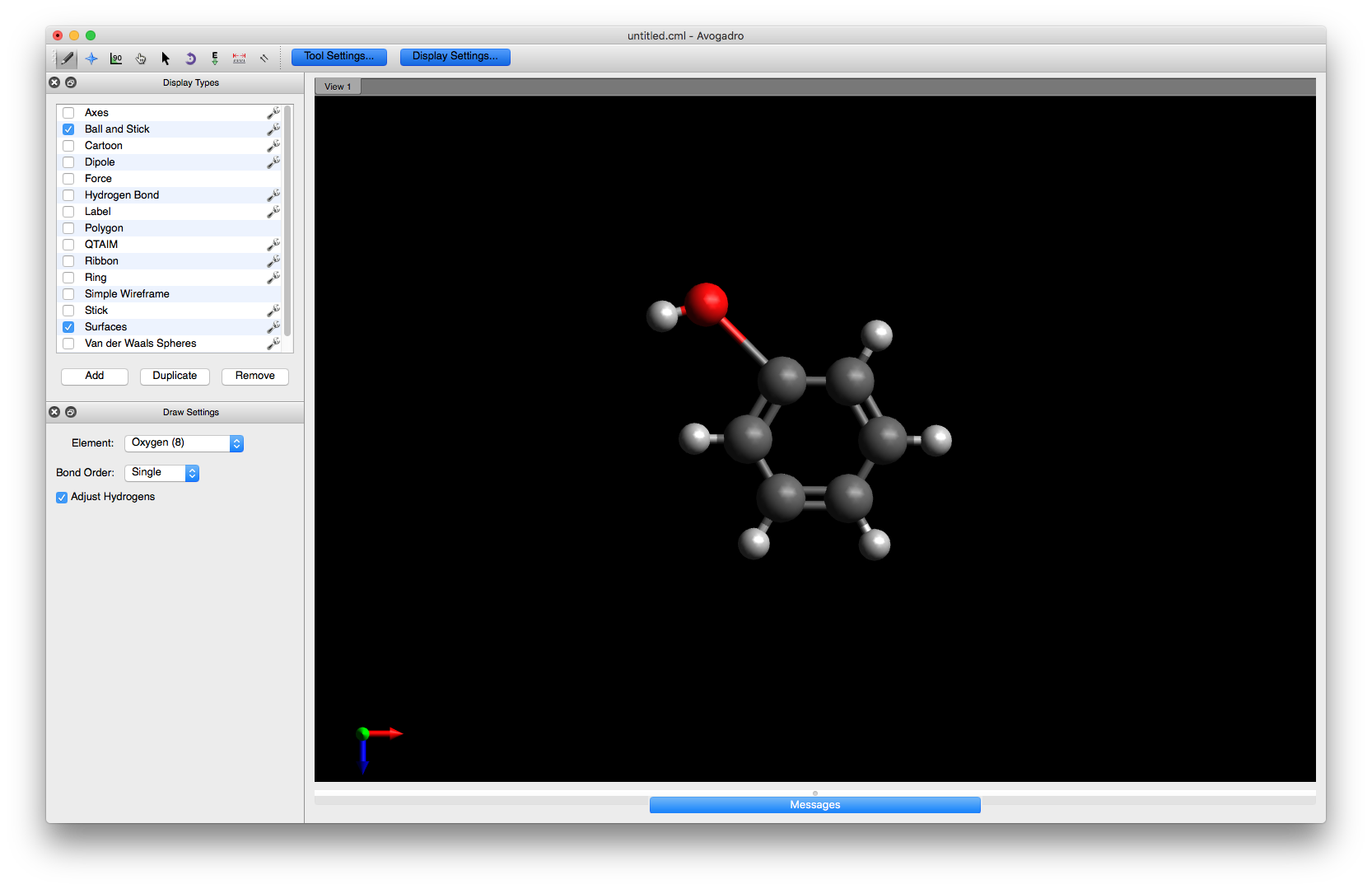
Note
If you make a mistake when adding/modifying atoms you can undo it using either the Edit -> Undo menu item or simply pressing the ctrl-z key combination.
Similarly, if you want to delete an atom or group you can select it with the mouse (using the Selection tool) and delete using Edit -> Cut or the ctrl-x key combination
Now optimise the structure with the MMFF94s force field as you did above and you will see the geometry of the hydroxyl group attachment improves.
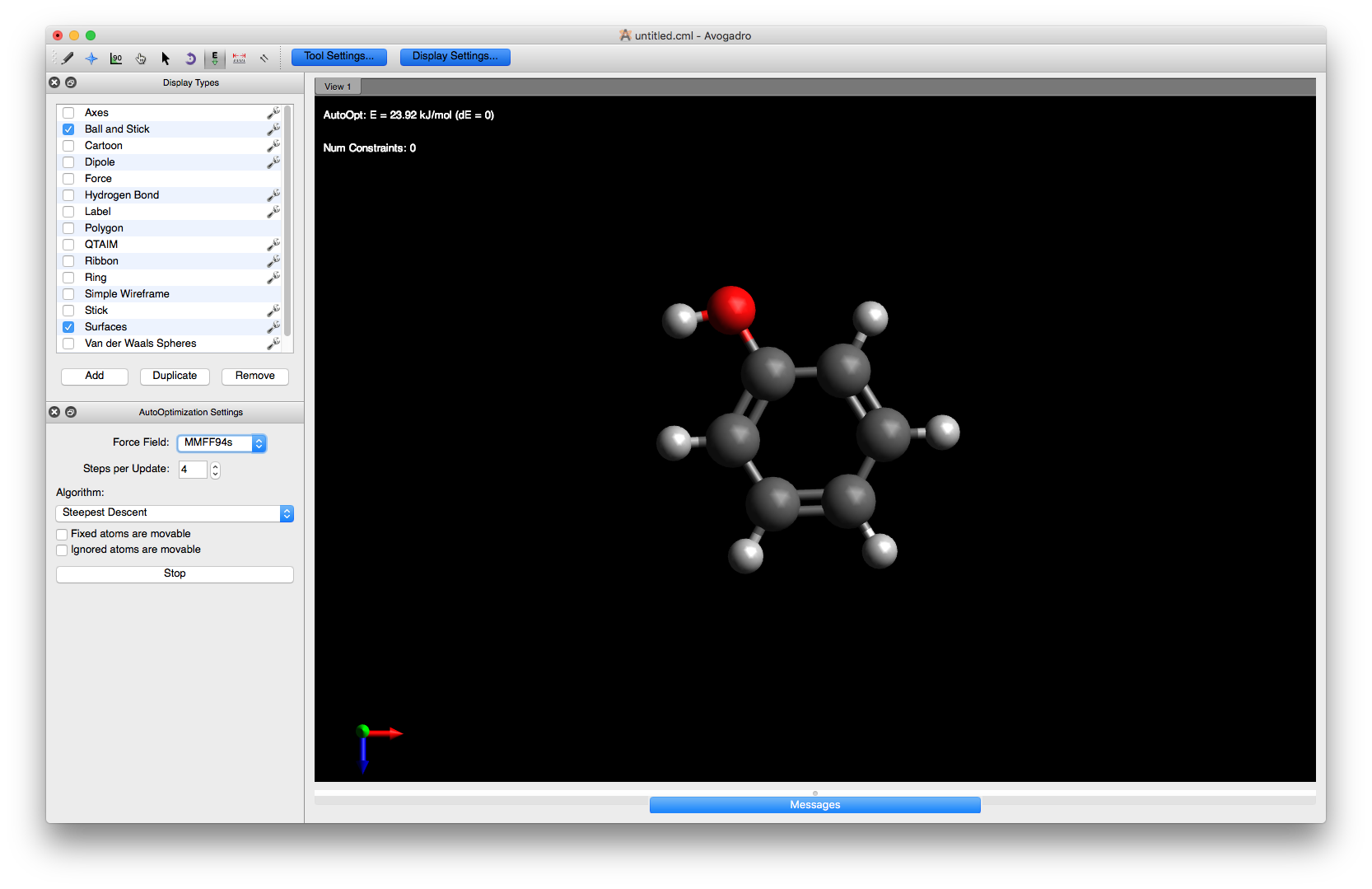
Note
Once you have prepared your ligand molecule you need to save it using some file format. Although the software we will be using to prepare the docking simulation, chimera, can handle a range of different file formats experience has shown that it is usually best to save your ligand(s) in mol2 format. This is because mol2 contains explicit information on the connectivity of the bonds and makes the conversion to pdbqt format for the actual docking run more reliable.
